微生物基因组重测序
产品名称: 微生物基因组重测序
英文名称: Microbial genome resequencing
产品编号:
产品价格: 询价
产品产地: null
品牌商标: null
更新时间: null
使用范围: null
- 联系人 :
- 地址 : 上海市浦东新区国际医学园区康新公路3399弄3号楼
- 邮编 : 201203
- 所在区域 : 上海
- 电话 : 159****9102
- 传真 : 021-51875086-8002
- 邮箱 : marketing@majorbio.com
产品介绍
微生物重测序能够快速解析菌株之间的变异信息。利用高通量测序技术,对一个物种的全基因组进行测序,并与近缘参考基因组进行比对,开发海量的SNP,InDel等分子标记,系统全面的检测菌株之间的基因变异信息。同时还可进一步进行物种进化、种群特征、选择压力等深入研究。
适用范围
有参考基因组的菌株或真菌;
选择具有代表性的个体。
技术优势
快速精准高效的解析基因组之间的差异,对全基因组的每一个碱基进行分析,获得最广泛的分子标记;
遗传变异类型丰富:单核苷酸多态性(SNP)、插入缺失(InDel)一网打尽,深度解析基因变异的各个方面;
快速高效:仅仅45个工作日即可完成全部分析内容,全方位加速微生物基因组快速发展;
精准分析:10年以上项目经验的专业分析人员深入解析,精确解决科研过程中的各个问题。
技术流程
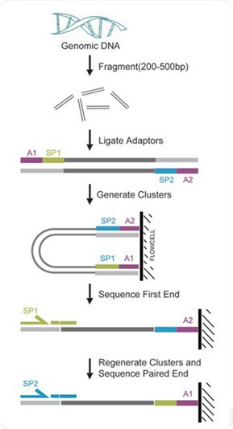
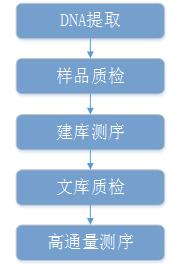
重测序建库及测序流程
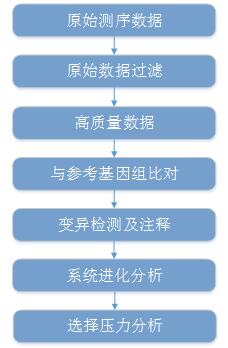
微生物重测序分析流程
|
解决问题 |
核心软件 | |
|
原始数据过滤 |
过滤低质量的序列 |
FastqQC/NGS-QC |
|
参考基因组比对 |
将测序reads比对到参考基因组 |
BWA/samtools |
|
多态性检测 |
研究菌株的地域传播规律 |
Samtools/GATK/BreakDancer |
|
变异基因功能注释 |
统计变异基因分布规律 寻找变异丰富的功能分类 |
Gene Annotation (In house)/Anovar/SnpEff |
|
系统进化树构建 |
探究不同菌株的进化关系 |
Mega/RaxML/PhyML |
|
选择压力分析 |
了解微生物进化过程 |
技术参数
|
产品 |
测序平台 |
测序策略 |
检测内容 |
项目周期 |
|
细菌重测序 |
Illumina PE150 |
350bp 文库 ≥ 100× |
SNP、InDel、SV |
45个工作日 |
|
真菌重测序 |
Illumina PE150 |
350bp 文库 ≥ 30× |
SNP、InDel、SV、CNV |
45个工作日 |
案例解析
案例一:蛭弧菌个体重测序
蛭弧菌(Bdellovibrio bacteriovorus)是一类能寄生于其它细菌,并能导致其裂解的革兰氏阴性菌。蛭弧菌比一般的细菌个体小,能通过细菌滤器,有类似噬菌体的作用,是一类能“吃掉”细菌的细菌。文章对一株野生型和一株突变型蛭弧菌进行了全基因组重测序,该突变型蛭弧菌可以不依赖宿主繁殖与生存。与参考基因组比对后,两株蛭弧菌分别获得28,386和28,379个突变位点,它们之间的差异突变位点为19个,其中包括一个2bp的InDel引起了hit基因的移码突变,该基因与蛭弧菌的宿主依赖性相关。
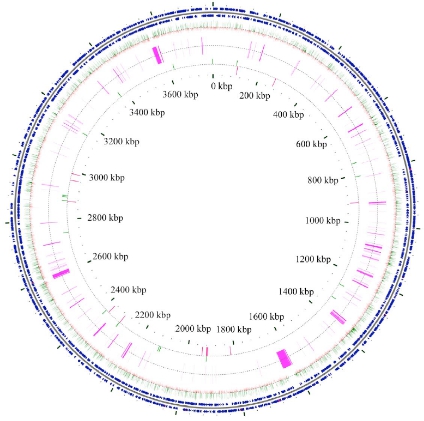
图1 两株蛭弧菌的基因组圈图
案例二:裂殖酵母群体重测序
裂殖酵母(Schizosaccharomyces pombe)是重要的真核模式生物,对100年来全球20个国家和地区收集的161个自然菌株的基因组进行了重测序,测序深度中值为76×。结果发现这些菌株有适中的遗传多态性(π=3×10-3)和较弱的群体结构。同时发现裂殖酵母的起源可以追溯到约公元前340年,并且一些重要的起源时间节点和古代文明的发展密切相关。通过全基因组关联分析揭示了基因型和表型变异之间的关系。
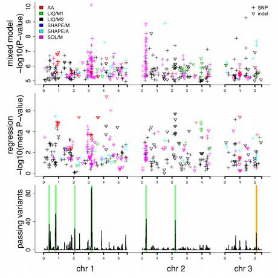
图1 全基因组关联分析结果
参考文献
[1]Wurtzel O, Dori-Bachash M, Pietrokovski S, et al. Mutation detection with next-generation resequencing through a mediator genome[J]. PLoS One, 2010, 5(12): e15628.
[2] Jeffares D C, Rallis C, Rieux A, et al. The genomic and phenotypic diversity of Schizosaccharomyces pombe[J]. Nature Genetics, 2015, 47(3): 235-241.
常见问题
(1)上机测序前如何判断样本是否有其他细菌污染?
送样后,美吉会对细菌样本的16S rRNA进行扩增、测序和比对,确认是否存在其他细菌污染,如果一代测序峰图有明显的双峰现象,或者NCBI的比对结果与预期结果不相符,则可以判断样本中存在污染。
(2)重测序分析是否进行de novo拼接?
能够比对到参考基因组上的reads序列是不进行de novo拼接的,但美吉会对没有比对到参考基因组上的reads进行de novo组装,发现基因组大片段插入和缺失。