微管蛋白β3单克隆抗体
产品名称: 微管蛋白β3单克隆抗体
英文名称: TUBB3
产品编号: hz-33177M
产品价格: null
产品产地: 中国/上海
品牌商标: HZbscience
更新时间: 2023-08-17T10:24:20
使用范围: WB=1:500-5000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500
上海沪震实业有限公司
- 联系人 : 鲍丽雯
- 地址 : 上海市闵行区闵北路88弄1-30号第22幢AQ136室
- 邮编 : 200612
- 所在区域 : 上海
- 电话 : 139****0749
- 传真 : 021-60345367
- 邮箱 : www.shzbio.net
Mouse Anti-TUBB3 antibody
| 产品编号 | hz-33177M |
| 英文名称 | TUBB3 |
| 中文名称 | 微管蛋白β3单克隆抗体 |
| 别 名 | Tubulin beta 3; beta III Tubulin; Tubb3; Tubulin beta-3; beta 4; MC1R; TUBB 3; TUBB 4; TUBB3; TUBB4; Tubulin beta 3 chain; Tubulin beta 4; Tubulin beta III; Tubulin beta-3 chain; Neuron-specific class III beta-tubulin; Syntaxin III; Neuron specific beta III Tubulin; Tubulin beta-4 chain; Tubulin beta-III; beta-4; CDCBM; CFEOM3A; M(beta)3; M(beta)6; Neuron-specific class III beta-tubulin; QccE-11995; QccE-15186; TBB3_HUMAN; Tubulin beta 4; Tubulin beta-4 chain. TUJ1 |
| 产品类型 | 内参抗体 |
| 研究领域 | 细胞生物 神经生物学 细胞骨架 |
| 抗体来源 | Mouse |
| 克隆类型 | Monoclonal |
| 克 隆 号 | 6F12 |
| 交叉反应 | Human, Mouse, Rat, |
| 产品应用 | WB=1:500-5000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复) not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 50kDa |
| 细胞定位 | 细胞浆 |
| 性 状 | Lyophilized or Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human TUBB3: |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein G |
| 储 存 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| 保存条件 | Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. |
| PubMed | PubMed |
| 产品介绍 | background: Neuronal Marker Beta III tubulin is abundant in the central and peripheral nervous systems (CNS and PNS) where it is prominently expressed during fetal and postnatal development. As exemplified in cerebellar and sympathoadrenal neurogenesis, the distribution of beta III is neuron-associated, exhibiting distinct temporospatial gradients according to the regional neuroepithelia of origin. However, transient expression of this protein is also present in the subventricular zones of the CNS comprising putative neuronal- and/or glial precursor cells, as well as in Kulchitsky neuroendocrine cells of the fetal respiratory epithelium. This temporally restricted, potentially non-neuronal expression may have implications in the identification of presumptive neurons derived from embryonic stem cells. Function: Tubulin is the major constituent of microtubules. Itbinds two moles of GTP, one at an exchangeable site on the betachain and one at a non-exchangeable site on the alpha chain. TUBB3plays a critical role in proper axon guidance and mantainance. Subunit: Dimer of alpha and beta chains. Subcellular Location: Cytoplasm, cytoskeleton. Tissue Specificity: Expression is primarily restricted to centraland peripheral nervous system. Greatly increased expression in mostcancerous tissues. Post-translational modifications: Some glutamate residues at the C-terminus arepolyglutamylated. This modification occurs exclusively on glutamateresidues and results in polyglutamate chains on the gamma-carboxylgroup. Also monoglycylated but not polyglycylated due to theabsence of functional TTLL10 in human. Monoglycylation is mainlylimited to tubulin incorporated into axonemes (cilia and flagella)whereas glutamylation is prevalent in neuronal cells, centrioles,axonemes, and the mitotic spindle. Both modifications can coexiston the same protein on adjacent residues, and lowering glycylationlevels increases polyglutamylation, and reciprocally. The precisefunction of such modifications is still unclear but they regulatethe assembly and dynamics of axonemal microtubules (Probable). Phosphorylated on Ser-172 by CDK1 during the cell cycle, frommetaphase to telophase, but not in interphase. This phosphorylationinhibits tubulin incorporation into microtubules. DISEASE: Defects in TUBB3 are the cause of congenital fibrosis ofextraocular muscles type 3A (CFEOM3A) [MIM:600638]. A congenitalocular motility disorder marked by restrictive ophthalmoplegiaaffecting extraocular muscles innervated by the oculomotor and/ortrochlear nerves. It is clinically characterized by anchoring ofthe eyes in downward gaze, ptosis, and backward tilt of the head.Congenital fibrosis of extraocular muscles type 3 presents as anon-progressive, autosomal dominant disorder with variableexpression. Patients may be bilaterally or unilaterally affected,and their oculo-motility defects range from completeophthalmoplegia (with the eyes fixed in a hypo- and exotropicposition), to mild asymptomatic restrictions of ocular movement.Ptosis, refractive error, amblyopia, and compensatory headpositions are associated with the more severe forms of thedisorder. In some cases the ocular phenotype is accompanied byadditional features including developmental delay, corpus callosumagenesis, basal ganglia dysmorphism, facial weakness,polyneuropathy. Defects in TUBB3 are the cause of cortical dysplasiacomplex with other brain malformations (CDCBM) [MIM:614039]. CDCBMis a disorder of aberrant neuronal migration and disturbed axonalguidance. Affected individuals have mild to severe mentalretardation, strabismus, axial hypotonia, and spasticity. Brainimaging shows variable malformations of cortical development,including polymicrogyria, gyral disorganization, and fusion of thebasal ganglia, as well as thin corpus callosum, hypoplasticbrainstem, and dysplastic cerebellar vermis. Extraocular musclesare not involved. Similarity: Belongs to the tubulin family. SWISS: Q13509 Gene ID: 10381 Database links: Entrez Gene: 10381 Human Entrez Gene: 22152 Mouse Entrez Gene: 246118 Rat Omim: 602661 Human SwissProt: Q13509 Human SwissProt: Q9ERD7 Mouse SwissProt: Q4QRB4 Rat Unigene: 511743 Human Unigene: 40068 Mouse Unigene: 43958 Rat Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| 产品图片 |
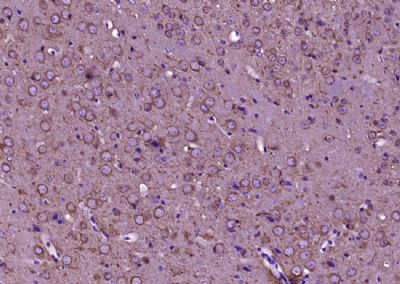 Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (TUBB3) Monoclonal Antibody, Unconjugated (bsm-33177M-1A2) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Mouse) (sp-0023) instructionsand DAB staining.
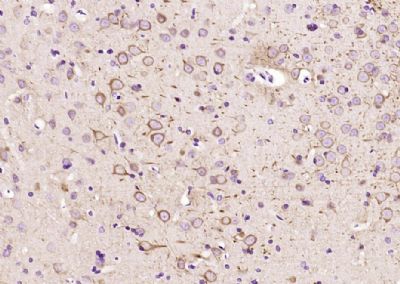 Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (TUBB3) Monoclonal Antibody, Unconjugated (bsm-33177M-1A2) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Mouse) (sp-0023) instructionsand DAB staining
|