16s rDNA测序
产品名称: 16s rDNA测序
英文名称: 16s rDNA sequencing
产品编号:
产品价格: 询价
产品产地: null
品牌商标: null
更新时间: null
使用范围: null
- 联系人 :
- 地址 : 上海市松江区九亭镇中心路1158号5号楼5层
- 邮编 : 200233
- 所在区域 : 上海
- 电话 : 400***0612
- 传真 :
- 邮箱 : market2@genenergy.cn
概述
16S rDNA测序,作为一种应用于环境微生物菌落多样性研究的靶标基因测序技术,通过针对覆盖16SrDNA的1个或多个连续可变性区域进行PCR扩增测序来鉴定样本微生物菌落成员和分析比较多样本微生物菌落的结构, 454测序平台以读长优势广泛应用于16SrDNA测序应用领域,但是Illumina的Hiseq2500和MiSeq平台可以针对16S rDNA的1个区域进行测序, 具有对样本更高的测序深度、鉴定更多的低丰度群落成员且以较低的费用的优势完成科研项目。
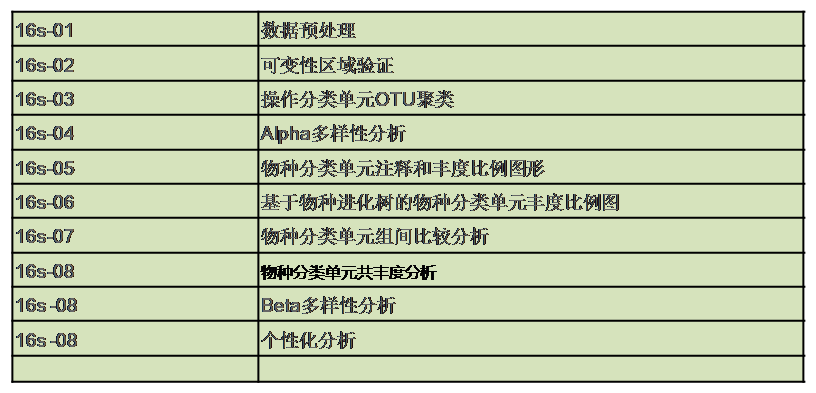
技术路线

生物信息学分析模块
样品要求
1. 样品总量:>=5ug
2. 样品浓度:>=50ng/ul
3. 样品纯度:OD值260/280应在1.8-2.0
4. 样品质量:基因组完整、无降解、无污染
5. 样品包装:用封口膜密封样品, DNA低温运输(-20°C)
参考文献
[1] J. Gregory Caporaso, Christian L. Lauber, William A. Walters, Donna Berg-Lyons, Catherine A. Lozupone, Peter J. Turnbaugh, Noah Fierer, and Rob Knight. Global patterns of 16S rRNA diversity at a depth ofmillions of sequences per sample.PNAS. 2010, Apr 30.
[2] Illumina Application Note.High-Speed, Multiplexed 16S Microbial Sequencing on the MiSeq System.
[3] J Gregory Caporaso,Christian L Lauber, William A Walters, Donna Berg-Lyons,James Huntley, Noah Fierer, Sarah M Owens, Jason Betley, Louise Fraser,Markus Bauer, Niall Gormley, Jack A Gilbert, Geoff Smith and Rob Knight. Ultra-high-throughput microbial community analysison the Illumina HiSeq and MiSeq platforms. 2012,Jan 1.
[4] Patrick H Degnan and Howard Ochman. Illumina-based analysis of microbial communitydiversity.The ISME Journal.2012, Jun 16.
应用案例
Applied Enviromental Microbiology. 2011, Apr 1.
Generation of Multimillion-Sequence 16S rRNA Gene Librariesfrom Complex Microbial Communities by AssemblingPaired-End Illumina Reads.
Andrea K. Bartram, Michael D. J. Lynch, Jennifer C. Stearns, Gabriel Moreno-Hagelsieband Josh D. Neufeld.
背景
1. 针对实验室培养的混合细菌系2个重复样本和来自北极苔原土壤2个重复样本利用Illumina 2×125 两端测序平台进行V3可变性区域测序,采用自己研发的两端序列重叠组装方法分别得到高质量的>1 million 和>6 million序列,生物信息学分析显示两个重复样本间高度相似性和证实可以million数量级序列检测到样本中的低丰度细菌种类。
2. 实验结果也提示可以利用多个标记和扩增16S rDNA不同可变性区域的引物序列能够同时检测多个样本的微生物菌落。
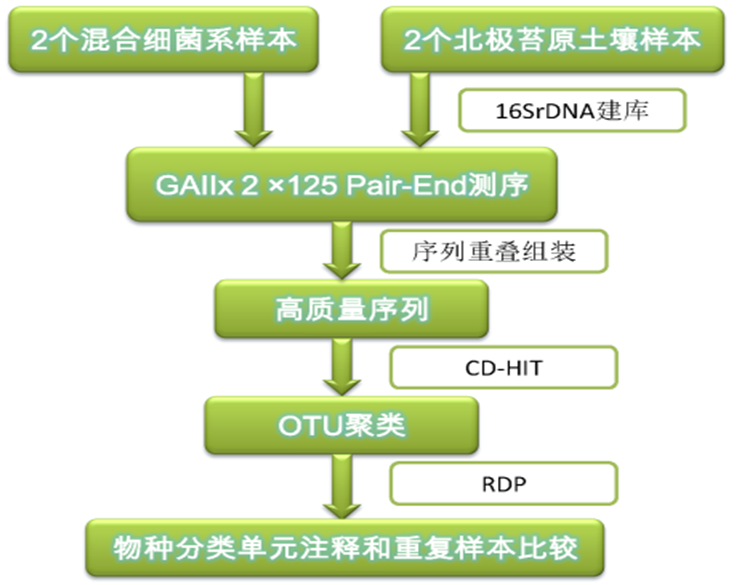
研究方法
1. 2个实验室培养的6个混合细菌系重复样本和2个来自北极苔原土壤重复样本分别抽提DNA
2. 修改后的341F和518R引物扩增V3区域,加标记接头, Illumina测序
3. 两端序列重叠组装
4. 序列的物种分类单元注释、操作分类单元OTU聚类和Good Coverage估计、
分析结果
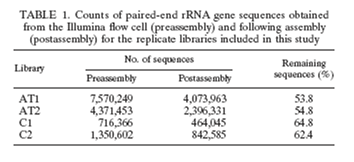
1.针对实验室培养的混合细菌系2个重复样本(C1、C2)和来自北极苔原土壤2个重复样本(AT1、AT2)序列重叠组装后的序列统计结果显示C1和C2共1,306,630条, AT1和AT2共6,470,294条。
2. 扩增V3区域测序列,两端序列重叠组装,然后利用OTU聚类和RDP注释得到在门注释水平上两个重复样本存在较高相似性, AT1和AT2相似性达到0.99, AT1和AT2分别与ATS(Sanger测序)达到0.95。
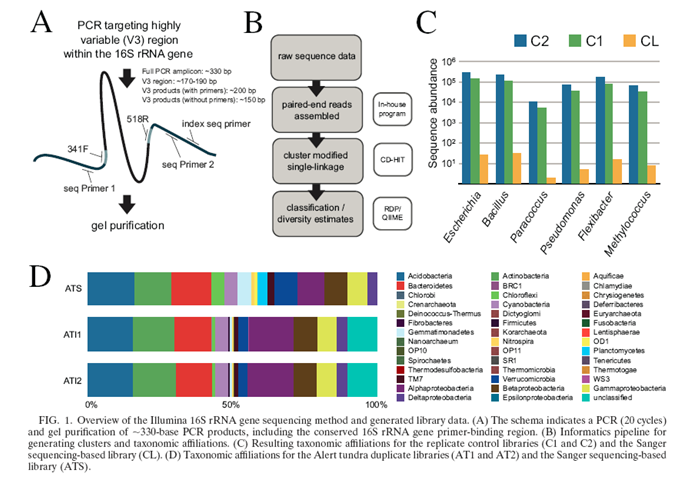
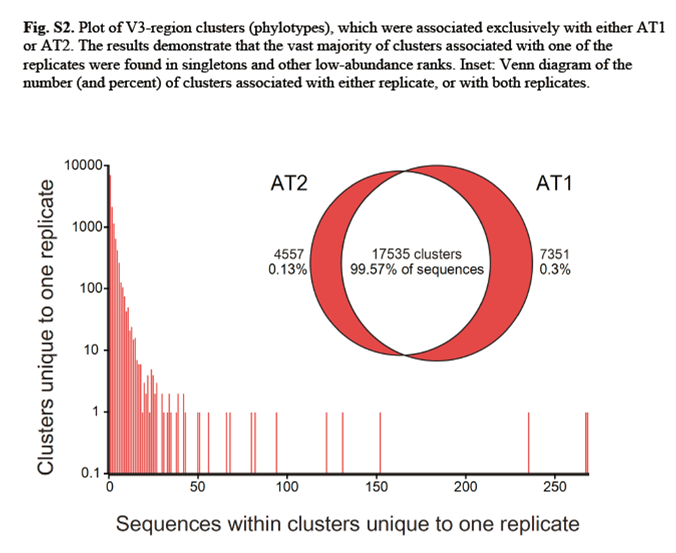
3.AT1和AT2样本的细菌种类交集大部分是高丰度细菌种类,T1和AT2样本特异的细菌种类大部分是低丰度细菌种类。
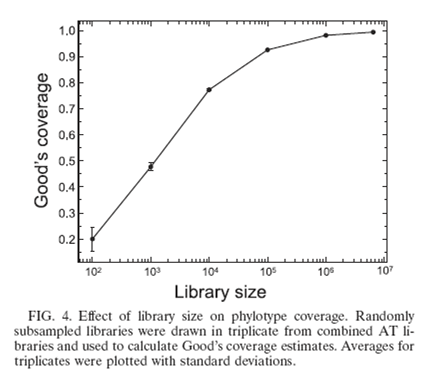
4.AT1和AT2两样本混合后的AT样本Good Coverage估计值曲线显示随着随机抽取的序列数增加,检测到的操作分类单元OTU数增加最后达到几乎饱和。